
Report on Annotation Standards
Version 1.0—June 2023
To accompany the Recommendations, the EBP provides A Report on Annotation Tools Recommendations.
AUTHORS: Fergal J. Martin, Françoise Thibaud-Nissen, Alice Denis, Roderic Guigó, Katharina J. Hoff, David Swarbreck, Jill Wegrzyn and the EBP annotation subcommittee
Overview
This document is provided by the EBP Annotation Subcommittee to give information and guidelines surrounding genome annotation, with a particular focus on gene annotation. It covers common approaches to annotation and provides guidelines for the use of transcriptomic and homology data, measuring annotation quality, and making annotation FAIR. It closes with a perspective on some of the major challenges facing genome annotation. The document is designed to be a high level overview of annotation, as opposed to an in-depth guide. We anticipate that detailed protocols for annotation of specific clades will be developed over the coming years and the document will be updated to include links to these as appropriate.
Introduction
High-quality annotation is required to transform reference genome sequences into actionable knowledge. In the most expansive definition, genome annotation is an accounting of the role and history of each basepair in the genome. However, for most eukaryotic species it is likely impossible to state that an annotation is complete, given the vast temporal and spatial complexity of the transcriptome and associated regulatory features. Moreover, for any annotation, completeness and accuracy is a function of the assembly quality and the available functional evidence.
A major aspect of genome annotation is the identification of genes. A variety of approaches exist for finding genes, ranging from direct prediction from the raw genome sequence, to utilizing data from other closely related species, to aligning same-species transcriptomic data. Each approach has its strengths and weaknesses and it is common for different approaches to be combined to produce the final gene set, as no single approach generally gives complete coverage of the structure of all genes.
Generating high-quality annotations is a complex process. Groups such as Ensembl at EMBL’s European Bioinformatics Institute (EMBL-EBI) and RefSeq at the NIH’s National Center for Biotechnology Information (NCBI) provide annotations across a wide range of species for which there is a publically available genome sequence (and in most cases associated transcriptomic data) and are focused on scaling genome annotation in line with the needs of the EBP. Many institutes and smaller groups also have experience with annotating genomes and often submit their annotations into the International Nucleotide Sequence Database Collaboration (INSDC) while submitting the corresponding genome assembly. In addition, a small but growing collection of research groups are actively working on critical problems in annotation including methods development, usability, and scalability. As a result, there are a significant number of available software packages for genome annotation. Here we will discuss some high-level aspects of genome annotation, with a focus on annotating genes.
Purpose and Audience
The aim of this document is to specify the features to be annotated on all EBP genomes and to provide guidance about the supporting evidence used in the creation of the annotation.
We seek to ensure that the annotation that is produced is appropriate given the assembly and available evidence, and that the annotation is comprehensively described based on the FAIR principles, so that it is as useful as possible for downstream analysis.
We recognise that, for any specific genome, annotation may be produced by multiple groups and there is no requirement for any group or methodology to annotate all or a large fraction of EBP genomes to meet this standard.
Annotation of Genomics Features
Genomes contain many different types of genomic features. A high-quality genome annotation will highlight a large array of genomic features, with accurate identification of feature boundaries and whenever possible functional annotation of features. For many species it will not be possible to generate a high-quality annotation, due to a lack of additional data outside of the raw genome sequence or other constraints such as a lack of available compute or domain knowledge.
Due to their importance to downstream analyses, gene annotation will be the focus of this document. Below we detail the minimal set of features that should be present in all genome annotations, in addition to listing the broader set of features that are expected when creating a high-quality genome annotation. Note that the list of features associated with a high-quality genome annotation are guidelines, not requirements; a high-quality annotation should represent several, though not necessarily all, of the feature types listed.
Genome features to be annotated in all genomes:
The EBP annotation standards committee proposes that the following feature classes are annotated in all genomes:
Repetitive regions, for the purpose of masking
Protein-coding genes:
CDSs
Useful and highly desired additional annotation:
Protein-coding genes
Predicted functional assignments
Non-coding RNAs (ncRNA):
rRNAs
tRNAs
Repeat elements (simple and transposable)
Classification through homology/structural assessment
CpG islands
Features associated with a high-quality genome annotation:
Protein-coding genes:
Transcription Start Sites (TSSs)
Untranslated regions (UTRs)
Promoters/enhancers
Pseudogenes
Long non-coding RNAs (lncRNAs)
Small RNAs (sRNAs)
microRNAs
snoRNAs
snRNAs
piRNAs
exRNAs
scaRNAs
Open chromatin regions (OCRs)
Accessible chromatin regions (ACRs)
Low complexity regions
telomere
centromere
Methylation
Immune genes
Variants including SNPs and structural variants
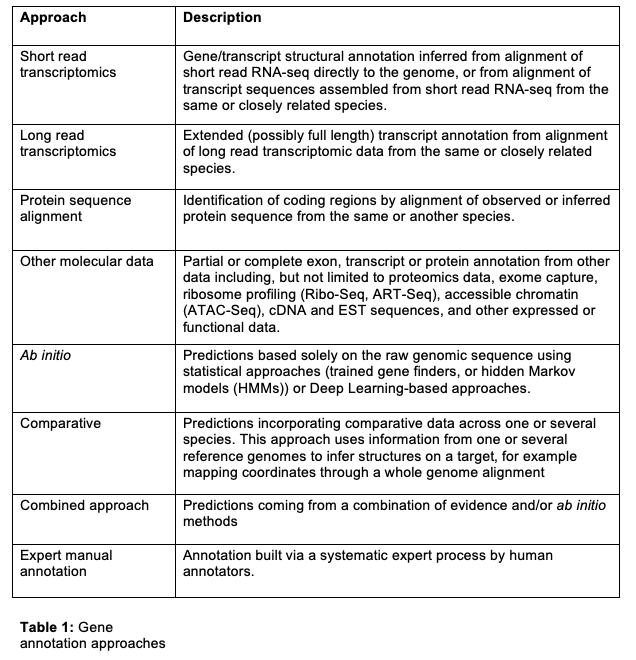
Gene annotation approaches
There are several approaches to the annotation of genes and transcripts. In general they are tightly linked with underlying evidence the approach uses. Ab initio annotation is the only approach listed that does not incorporate additional evidence, and instead is based purely on the genome sequence. All other approaches use external evidence, which may be same-species or cross-species in origin to help build a structural annotation of the transcripts. Note that combined approaches are common, i.e. parts of a single transcript may be annotated using support from short read transcriptomics, while other parts of the same transcript may be predicted ab initio. Combined approaches are encouraged, as each individual approach has limitations. Table 1 shows a list of the major approaches.
6. Guidelines on the use of evidence for annotation
The use of same-species or cross-species data results in a higher-quality annotation than a pure ab initio approach. The quality of the final annotation is strongly correlated to how small the evolutionary distance is between the sequence of the evidence and the sequence of the target genome. The more similar a sequence is to the genome, the more accurate the alignment and resulting annotation will be. As such, same-species evidence, particularly short and long read transcriptomes, leads to the higher-quality annotations. UTRs, for example, can only be accurately predicted using same-species transcriptomics data. Below we describe some guidelines for the use of evidence in annotation.
We strongly encourage using supporting evidence originating from long standing and well trusted sources, for example the Sequence Read Archive (SRA)/The European Nucleotide Archive (ENA) for transcriptomic data, UniProt/RefSeq/Ensembl for protein sequences or RefSeq/Ensembl for transcript sequences.
Use of transcriptomic evidence
The completeness and quality of a gene annotation is substantially higher when it incorporates transcriptomic data from the same species (ideally the same individual that has had its genome sequenced). For this reason, we recommend the collection of transcriptomic data at the same time material is collected for the genome sequencing. Indeed, some annotation pipelines require such data.
Although the optimal quantity and quality of transcriptional data to support annotation is highly dependent on the organism and the quality of the libraries, broad statements can be made:
Sequencing from a diverse collection of tissues and conditions will provide a more complete catalog of expressed genes than sequencing from a narrow set. For vertebrates, tissues such as the brain, gonads and lung/gill have the most diverse transcriptomes and thus should be prioritized, while liver/muscle/blood generally show the least diversity. For eukaryotes, sampling from as many tissues and developmental stages as possible is better, in particular sampling tissue types known to have complex expression profiles. For very small organisms, whole organism samples can be used.
Short RNA-Seq reads in the 100 to 150 bp range, such as those generated with Illumina, are valuable for their depth and for their sequence accuracy. The most common approach for annotating using short reads is to map the reads to the genome using a short read spliced aligner and then infer the corresponding transcript structures using transcript reconstruction software. The limitation of short reads is that the transcript structures are generally inferred as opposed to directly observed, so the resulting structures may not be biologically real. For protein-coding genes, assessment of the ORF against known proteins can help measure confidence. De novo assemblies of short reads to generate synthetic full-length cDNA are computationally expensive and error prone, and are therefore not recommended.
In general, for short read data, the higher the number of reads the better. Experience in RefSeq and Ensembl shows that ideally individual tissues will have ~200 million reads each, with 5 or more tissues/development stages being preferable. However, this is highly subjective and will vary from species to species. In practice, this is often not feasible, but even a smaller total number of reads (e.g. 50 million) can substantially improve the quality of an annotation.
Long read transcriptomes (such as consensus PacBio reads or Oxford Nanopore) are a useful complement to short reads because the long range information allows for more accurate identification of transcript structure and diversity. However, long reads, particularly ONT data, currently have a higher error rate than short reads, and libraries are not sequenced as deeply as short read libraries. This can have a significant effect on structural accuracy of annotation. As a result, it may be necessary to combine long reads with other evidence sources (e.g. overlapping short read mappings) to refine intron/exon boundaries, and remove artifacts. The cost/benefit ratio of high-quality full-length cDNA sequencing versus short reads is evolving rapidly and will need frequent re-evaluation.
Use of evidence data from other species
Evidence data from other species (i.e. homology information) is commonly used. The value of this information is a function of the type of information and the sequence divergence between the target genome and the source of the evidence.
Aligning proteins from closely related species to the genome in a splice-aware manner is a common approach that can yield good results if the proteins used provide a high coverage of the proteome of the species and the evolutionary distance is not large. A low divergence rate can yield accurate structural annotation, with small exons, particularly terminal exons being most problematic. In the case of hint-guided gene prediction, protein-to-genome alignments can be used over much greater evolutionary distances as they are generally used to help constrain predictions of coding exons on the genome as opposed to explicitly defining structures. Regardless of the approach used it is important that the protein sequences come from trusted sources such as UniProt entries with associated experimental evidence, RefSeq NPs, or proteomes derived from an accurately annotated reference.
Whole genome alignment with a suitable reference genome (see ‘Comparative’ in Table 1) is a computationally expensive, but often accurate approach to mapping the coordinates of protein-coding exons. This generally involves a pairwise alignment between the reference and target genomes, the recovery and chaining of alignment blocks and the subsequent mapping of coding exons. This approach can work well even at reasonable levels of nucleotide divergence due to the long range context provided by the alignments. It can also deal with features such as mapping small exons.
Cross-species nucleotide data such as predicted transcript sequences, and long/short read data can be used to annotate target genomes via splice-aware local alignment. However, even relatively modest levels of divergence (past 5 percent), can result in alignment errors that become more pronounced as the divergence increases. In addition, some of the associated tools may need to be fine tuned to allow increased divergence, as many work off k-mers and are defaulted to expect near exact matches between the transcriptome and genome. Similar to protein data, divergence may be more tolerable when used in a hint-guided gene predictor as opposed to just direct alignment to the genome.
In the above cases it is recommended to calculate the percent coverage and identity between the sequence of the original evidence and the sequence annotated on the target genome, as this can give an indication of the quality. These values can then be used to filter out low confidence alignments/structures.
7. Estimating Annotation quality
For the foreseeable future, annotation quality will be measured in a relative scale as compared to other genomes within the same clade. Near complete genome annotation is unlikely to be possible for nearly all species. When the quality of annotation for given species has fallen substantially below the relative level for similar species, it should be prioritized for reannotation.
To facilitate comparison and evaluation, basic statistics on the gene set should be provided including the number of genes per major class (protein-coding, long non-coding, small non-coding, pseudogene). Within each of the classes annotated, basic information on the observed structures should be provided. This includes average number of transcript per gene, average transcript length, average number exons per gene, average number of CDS exons and CDS length (where applicable). In addition to basic metrics on the observed structures, potential structural abnormalities or methodological issues should be recorded. This includes missing CDS start/stops, non-canonical splicing, over estimates of mono-exonic genes, frameshifts or abnormally small introns (< 50bp).
Quality of the annotation should be estimated using Benchmarking Universal Single-Copy Orthologs (BUSCO), which calculates the presence/absence of single-copy marker genes in a genome or an annotated protein set. The application should be run in protein mode for the annotated protein-coding genes, selecting a representative transcript per gene (typically the transcript with the longest CDS). The results may be compared to results obtained in genome mode. As the completeness of a particular BUSCO marker set varies significantly from clade to clade (and depends on the level within the taxonomy), where possible it is useful to present metrics in the context of other species and annotations estimated using the same set of markers. Newer tools, leveraging more taxonomically diverse reference data and/or machine learning are emerging and may be considered as well.
Other approaches for evaluating the quality of an annotation, such as calculation of reciprocal best hits or orthologous genes to a reference species, may be used. Results will be more valuable to consumers of the annotation if provided in the context of multiple species, and if reproducible with a stand-alone software package.
8. Making annotation fair
The Earth BioGenome Project is committed to developing a network of communities able to navigate from biological samples to genomes, to gene sets, and to multi-species analysis products. Annotation providers both receive and produce data and are therefore at the nexus of this web. As such, they should strive to make their data easy to find, download, use in downstream applications and reuse by others, by adhering to FAIR principles (Findable, Accessible, Interoperable and Reusable). We detail below our views on best practices to produce FAIR annotations.
Findable
Annotation providers must provide identifiers and proper metadata that can be unambiguously used in scientific publications and communication, and that make the annotation as whole, and subsets of features findable. In particular, the annotation as a whole should have an identifier (e.g. GCF_025698485.1-RS_2022_12), and be associated with a specific genome assembly (e.g. GCA_025698485.1), an annotation provider, a date, and a short phrase for the pipeline or method used.
Individual features annotated on the assembly should also have identifiers (e.g. At1g79570) and be associated with a class (coding gene, CDS, UTR, pseudogene, repeat, etc.), and as much as possible have an associated function. Supporting evidence, orthology to genes in other species, and confidence level in the feature prediction are also encouraged.
Accessible
All annotations must be freely available without any restrictions on further use (equivalent to public domain or cc0 licensing).
The Ensembl and RefSeq groups may only annotate genomes after they have been submitted to an INSDC database (GenBank, ENA, the DNA Data Bank of Japan), have been accessioned with an GCA_xx identifier, and have been publicly released. Supporting transcriptome data for annotation must also be submitted, accessioned, and released via a recognized archival database before it will be used by central annotation services. Such databases currently include ArrayExpress, the Gene Expression Omnibus, the SRA and INSDC.
As much as possible, annotation produced by other groups should be submitted to an INSDC archive, so it can benefit from the archive’s accessioning and indexing infrastructure and so that the annotated features can be searchable by name, accession or sequence.
Additional groups proposing themselves as central annotation services in the context of EBP would be expected to join the genome browser agreement and meet the requirements of this section.
Interoperable
Annotation must be provided in standard file formats such as GFF3 with standard descriptive attribute tags. This is an area of active development and we expect future versions of this standard to specify a specific dialect of GFF3. This dialect will likely include, among other specifications, specific attribute tags in the future for information such as alignment, protein domain, gene family, and other information. To the extent possible, we intend to collaborate with other efforts to standardize GFF3 usage including the EBP IT and informatics standards committee.
Reusable
To bolster confidence and maximize re-usability, we encourage annotation groups to document publicly the genome, software components, input data, details of the process that was used to produce the annotation, as well as the quality metrics of the gene set. The genome assembly must be published in an INDSC database and referred to by its accession (GCA_*), as assembly names are not guaranteed to be globally unique and are subject to change. The annotated features must be mappable to sequence records in the assembly, although the provider may use different nomenclature for a given sequence (eg. chromosome 1) as long as the corresponding INSDC sequence identifier is incorporated into the annotation release. The software components, and pipeline should preferably be published and the exact versions provided.
Identifiers for the evidence data used as input for the prediction of genomic features should be listed. This could be in the form of sample accessions for INSDC records or release numbers/accessions for Ensembl/RefSeq/UniProt/AGR, or in the form of a query used to retrieve data from a database with the date on which this query was executed. If the annotation was derived from other annotated genomes by comparative methods the accessions and annotation identifiers for these genomes must be provided.
The use of private data is discouraged as it limits the ability for others to understand the fine detail of the annotation.
9. Key challenges for improving genome annotation
We close this version of the standard with a short perspective on what is needed to improve annotation. This is a limited list of areas that should be targeted for further investment by funding agencies. We note that overall investment in genome annotation has been relatively low over the past 15 years as many computational groups have focused on other research problems such as short read and next-generation sequencing informatics including tools for the analysis of RNA-seq and single cell sequencing experiments. As EBP and other projects begin to produce many more high-quality genome assemblies, there are considerable opportunities now to improve our collective ability to annotate these genomes and thus learn as much as possible about their biology.
Below are some of the major challenges and opportunities that need to be addressed in regards to genome annotation.
A community effort to build a standardized, cloud native, annotation pipeline. The genome assembly community came together to work on a generalized pipeline for genome assembly and this led to many of the most active members of that community communicating on a regular basis to create a workflow for genome assembly. This allowed for rapid progress on building high-quality genome assemblies, and is something that should be replicated in the annotation community. While it is not expected that a single annotation pipeline would work for all species, having a base workflow that can be tailored as needed to specific species/clades and bringing people together to work on this would likely drive other improvements in genome annotation.
Linked to the above, there is a need for more active development in regards to the tools used in annotation. While there have been some notable advances in tools over the past few years, there is still a relatively limited number of groups working on annotation tools, and many commonly used tools are very old, with some no longer being actively worked on. If the annotation community can come together to build standardized workflows, this will also highlight areas where the tool chain is insufficient and incentivize development of replacement tools. This is a process that happened in the genome assembly process, where there have been many tools developed to solve issues identified during the creation of the workflow.
There is a great need for metadata standards, file format standards, versioning and annotation quality metrics to be formalized for annotation. Currently there is little standardization in any of these areas. Recording the evidence and pipelines used to create an annotation is critical for comparison between annotation and understanding issues that may be present in the annotation of a particular gene. For storing annotations, GFF3 may be the de facto standard, however the specification allows for a high degree of variance between annotations created by different groups. In terms of assessing annotations, we have already discussed BUSCO, which is the preferred community option for judging annotation quality, however there are many clades for which the BUSCO sets are not comprehensive, and this approach is simply one method for understanding quality. Having a robust and agreed upon set of metrics, which could include things like BUSCO scores, will be important for understanding and comparing the relative quality of genome annotations.
Training a new generation of annotators is essential to the future of genome annotation. As tools and technologies improve, annotation naturally becomes more of a black box. However, now more than ever it is critical that existing members of the annotation community pass on domain knowledge to the next generation. There will still be a need for manual examination and correction to annotations, particularly for key genes in species with a high scientific or commercial interest. Manual annotation is also highly valuable for improving the quality of potential training set for machine learning approaches. On the other side, there is also a need for training existing members of the community in areas such as creating containerized pipelines and deploying them on the cloud.
The take home message from the above is that it is important that the community comes together to work on shared problems and create new standards to push the field of annotation forward. This effort should include large annotation hubs such as RefSeq and Ensembl, but also the many individual groups that are carrying out annotation across the globe. For this to happen, there is a need for investment in annotation, to help build new tools and workflows and ensure the process can be scaled to the needs of the EBP. Without accurate genome annotations the value of these high-quality genome assemblies will not be fully realized.

About the Subcommittee
This Report on Annotation Standards was developed by EBP’s Scientific Subcommittee for Annotation.